


网页端用户可点击右方目录直接跳转至制定章节
转发本文到朋友圈,添加下方二维码,免费领取【OOS调查记录】
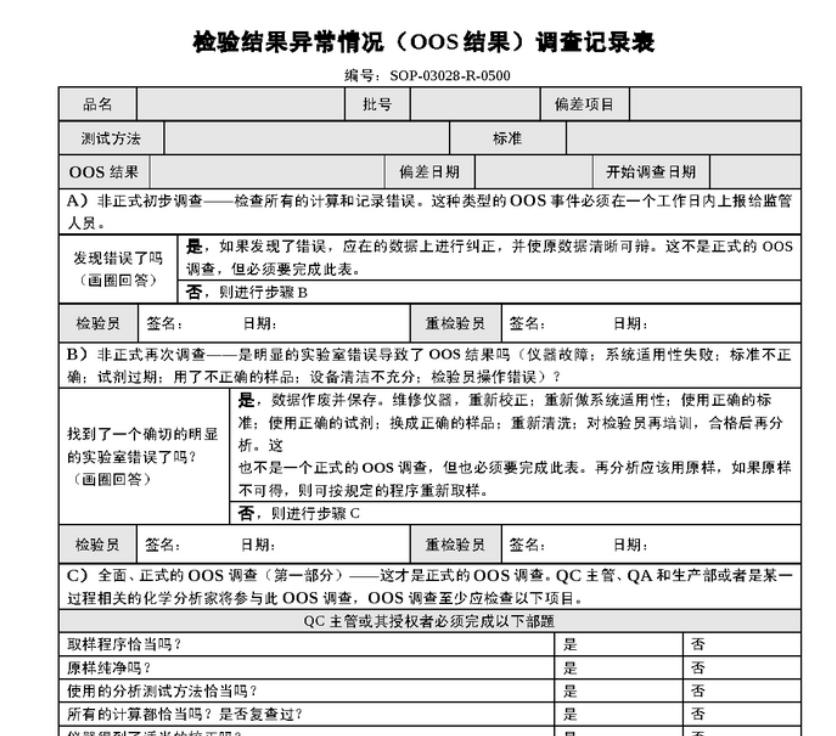

1. 概述
超标(Out of specification, OOS),结果超出设定质量标准。其中包括注册标准以及企业内控标准。如果对于产品有多个接受标准,结果的评判采用严格的标准执行。 当两份平行测试样品检验结果一份合格、另一份不合格时,不得将其平均,亦应视为超标。
一旦出现超标或超趋势的结果,必须进行实验室调查以便确认结果是否有效。即使已根据确认有效的超标结果判定一批产品为不合格品时,仍需进行调查以找出确切的或可能的不合格原因,并评估该产品或其它产品的其它批次是否受该超标结果的影响。 在调查过程中,应对发现的任何错误采取相应的预防和整改措施。
1.1 关键概念辨析
超趋势(out of trend, OOT):
结果虽在质量标准之内,但是仍然比较反常,与长时期观察到的趋势或者预期结果不一致。如,某成分含量(标准:96.0%-103.0%)历史上典型值为98.0%-101.0%,但此次测定结果为96.8%或102.3%,即构成一次超常检验结果。典型值即OOT限度是需要文件指导去制定的,检验结果超出限度即认为为OOT结果。
OOS结果:在检验过程中出现的任何偏离标准(包括药品申报标准、DMF中申报标准、药典标准和企业内控标准)的结果。OOT结果:超出了正常波动范围但未偏离任何标准。两者的本质区别在于是否偏离标准。
一般来讲,OOT的限度应由企业根据以下原则制定:
异常数据(abnormal data, AD):
指超标(OOS)及超趋势(OOT)以外的异常数据或来自异常测试过程的数据或事件。
实验室调查是明确出现数据偏差的原因,避免不合格品或潜在风险产品流入市场。对于上述任何超出质量标准及趋势或异常的分析结果,企业都须进行实验室调查。此调查应当遵循真实、科学、有效的原则,且必须符合相应的法规要求。
复验(Retest):
也称原样复验或再检验,指对同一次所取的样品的一部分按规定方法重新制备,进行化验。注意不要使用制备好的样品,应由另一经验、资历更为丰富的检验员完成而不是原检验人员。
目的:调查00S结果是否由检验用仪器设备或系统故障、操作程序错误或操作失误等实验室错误造成的,而不是复核或对第一次检验结果的纠正,也不能认为复验的结果就一定正确。
再取样(Resample):
再取样或重新取样,指按照规定的取样程序,对发生00S批次的产品重新进行取样,获取供检测的新样品。
再取样并不是随意决定的,需要有合理理由并经批准,并且记录下来。
分析错误:
指造成分析结果与真值的偏差的原因,是实验进行中的错误。
微生物数据偏差(microbial data deviation,MDD):
国际上的监管指导原则一般将OOS的焦点集中于分析化学检测,对微生物实验室出现的不合规范结果讨论较少。美国FDA的一个微生物试验专门工作组首次于2001年提出的一个首字母缩略词MDD,将微生物实验室的数据偏差与化学检验项目的OOS区别开来。
MDD包含检验过程出现与微生物相关的所有不合规范的数据,不仅包括终产品的结果偏差,也包括过程控制中的偏差。MDD 调查可分为两个阶段:一是聚焦于 MDD 是否无效,二是在 MDD 有效的前提下开展全面调查。MDD 调查包括但不限于实验室环境、抽样区的防护条件、样品在该检验条件下以往检验的情况、样品本身具有使微生物存活或繁殖的特性等情况。 此外,还应回顾试验过程,也可评价该实验结果的可靠性及实验过程是否恰当。
2. 法规指南
2.1 国内药监
( 中国GMP 通则)
中国GMP 通则)
中国GMP 通则)第十章 质量控制与质量保证
第二百二十四条 质量控制实验室应当建立检验结果超标调查的操作规程。 任何检验结果超标都必须按照操作规程进行完整的调查,并有相应的记录。
2.2 FDA
Sec. 211.160 一般要求
(a)对本子部所要求制订的所有质量标准、标准、取样方案、检验规程或其它实验室控制机制,包括这些质量标准、标准、取样方案、检验规程或其它实验室控制机制的变更,应由合适的部门起草,并由质量管理部门审核和批准。应遵循本子部的要求并即时做好记录。所有偏离既定质量标准、标准、取样方案、检验程序或其它实验室控制机制的偏差,均应记录并作出合理解释。
Sec. 211.165 检验与发运放行
(d) 由质量管理部门使用的取样和检验的验收标准,应能充分保证各批药品符合适当的质量标准和适当的统计质量控制标准,以此作为批准和放行的条件。统计质量控制标准应包括适当的接受水平和/或适当的拒收水平。
(f) 不符合制订的标准或质量标准或其它相关质量控制标准的药品应予拒绝。可采取返工。在接受和使用前,返工物料必须符合适当的标准、质量标准和任何其它相关的标准。
Sec. 211.192 生产记录审核。
在批放行或发运之前,所有药品生产与控制记录(包括包装与贴签)都应由质量管理部门进行审核和批准,以确定与所有既定的批准的书面规程的符合情况。不管该批是否已经发运,所有未解释的差异(包括超出生产与控制主记录中设定的最大或最小限度的理论收率)或一批或其任何原辅料不符合质量标准的情况,都应进行彻底调查。调查应扩大至相同药品的其它批次和与差异或不合格情况相关的其它药品。调查应作出书面记录并应包括结论和追踪情况。
3. 鉴定和评估 OOS 检测结果 — 第一步:实验室调查
FDA 法规要求当获得 OOS 结果时应展开调查。调查的目的是确定OOS结果的原因。应确定是测量过程的异常还是生产工艺异常是 OOS 结果的来源。即使一个批次因 OOS 结果被拒绝,调查仍是必须的,以确定该结果是否与同种产品其它批号或其它产品有关,批次拒绝不能否定实施调查的需要。法规要求要做调查的书面记录,包括结论和跟进措施。
4. 调查 OOS 结果 — 第二步:全面的OOS 调查
如果初步评估并不确定是实验室错误引起的 OOS 结果,检测结果显示准确的话,应开展使用既定规程的全面 OOS 调查。该调查可能包括生产工艺回顾和/或附加的实验室工作。此类调查的目的应是确定OOS 结果的根本原因并采取适当的纠正和预防措施7。一个全面的调查应包括对生产和取样程序的回顾, 并且经常包括附加的实验室检验。这样的调查应有最高的优先权。在这一阶段的内容中,是对 OOS 结果 对已销售批次的影响的评估。
5. 调查结论
A. 调查结果解释
质量控制部门应负责对调查的结果进行解释。初步的 OOS 结果未必表示该批次一定是不合格并且被拒收。应该对 OOS 结果进行调查,调查中的发现,包括复试结果,应加以解释以对该批次作出评价,并得出放行或拒收的决定。
对于调查揭露了原因的情形,并且可疑数据是无效的,那么该结果不应用于评估该批次的质量。只有依据可以合理确定引发了OOS结果的检测活动中观察到的现象与文件记录可以确定离散的检测结果是无效的。
对于调查显示OOS结果是由影响批质量的一个因素所导致的(也就是说 OOS 结果被证实了)情况,结果应该用来评估批次的质量。一个经确证后的 OOS 结果表明这批不满足既定的标准或规格,应造成批次被拒收并进行恰当的处理。对未得出结论的调查——即调查(1)没有表明 OOS检测结果的原因和(2)没有证实 OOS 结果——OOS 结果应在批处理决定中被充分考虑。
在第一种情况下(OOS 结果被确定),则调查应从一个 OOS 调查变成一个批次失败的调查,此时,调查必须扩展至与该具体失败相关的其他批次或产品。
在第二种情况下(无定论的),QCU 可能最终还是会决定将该批放行。例如,公司可能会在下列情景下考虑放行该产品:
一个产品有一个可接受的化合物检测范围 ,从90.0-110.0%。首次OOS 检测结果为 89.5%,随后由初始样品得到的样品得到下列复试结果:99.0%, 98.9%, 99.0%, 99.1%, 98.8%, 99.1%, 99.0%。实验室综合调查(第一阶段)并未发现任何实验室错误。对该批生产的回顾活动发现无异常或异常工艺波动14。对生产工艺的审核及该产品的历史表明工艺是稳定的。7个合格的复测结果都在所用方法的已知变动限度内。过程中监控、含量均匀度、溶出度和其他检测的批次结果均与合格的复测结果相吻合。经过彻底调查后,公司的质量控制部门可能认为首次 OOS结果不能反映该批产品的真实质量。
此类设想中值得注意的是,初始的完整实验室调查未发现任何可归结原因。尽管如此,如果接下来的调查得出结论 OOS 结果的原因与生产工艺不相关,对于这样检测实验室偏差的非典型失误,调查包括跟踪措施或仔细检查以防止可能导致OOS结果的实验室错误再次发生是非常必要的。
正如上述例子说明的,任何放行该批的决定,即使首次OOS 结果并未确认无效,应该再彻底的调查已证明该 OOS 结果并不反映该批的质量之后做出。在做这样的决定时,质量控制部门总是应谨慎。
5. FAILURE (OUT-OF-SPECIFICATION) LABORATORY RESULTS
Evaluate the company's system to investigate laboratory test failures. These investigations represent a key issue in deciding whether a product may be released or rejected and form the basis for retesting, and resampling.
In a recent court decision the judge used the term "out-of-specification" (OOS) laboratory result rather than the term "product failure" which is more common to FDA investigators and analysts. He ruled that an OOS result identified as a laboratory error by a failure investigation or an outlier test. The court provided explicit limitations on the use of outlier tests and these are discussed in a later segment of this document., or overcome by retesting. The court ruled on the use of retesting which is covered in a later segment of this document. is not a product failure. OOS results fall into three categories:
-- laboratory error
-- non-process related or operator error
-- process related or manufacturing process error
A. LABORATORY ERRORS
B. LABORATORY INVESTIGATIONS
C. FORMAL INVESTIGATIONS
D. INVESTIGATION DOCUMENTATION
E. INVESTIGATION TIME FRAMES
III. 报告质量数据和计算质量量度
C. 可以报告的质量量度数据
(2)无效的OOS率数据(IOOSR):
药品成品或API的批次放行和长期稳定性检验OOS结果个数。长期稳定性检验是用于支持标签标示的有效期的检验
药品成品或API的批次放行和长期稳定性检验总个数。长期稳定性检验是用于支持标签标示的有效期的检验
药品成品或API的批次放行和长期稳定性检验的OOS结果中,其来源被确认为检验过程过失的个数。长期稳定性检验是用于支持标签标示的有效期的检验
IOOSR数据具体标准:
得到任何OOS结果,必须开展调查。31在质量量度计划的范围内,以下OOS结果应当计入:(1)只包括药品成品、API放行检验和长期稳定性检验,(2)所有的药品成品和API以及长期稳定性检验结果,只要一开始显示出OOS的,都需要计入OOS结果,尽管OOS的来源经调查后被确定为检验过程中的失误。参见"FDA药品生产中OOS调查结果指南"(Investigating Out-of-Specification (OOS) Test Results for Pharmaceutical Production)(2006年10月)的第III部分,以及"无菌工艺生产无菌药品——CGMP"指南(Sterile Drugs Products Produced by Aseptic Processing – Current Good Manufacturing Practice)(2004年9月)的第XI部分
总检验个数数据作为一个测量手段可用于:(1)为确认无效的OOS率量度提供上下文背景,(2)作为衡量生产表现和产品符合标准能力的二级量度(批次放行和长期稳定性的OOS结果,最终调查认定是生产过程过失的个数,除以同一报告期内批次放行和长期稳定性的检验总数)
在质量量度计划的范围内,OOS结果应当在检验结果完成之日计入,或OOS调查开启之日计入
一个检验包括批次放行或一个稳定性时间点的单个检验结果及其对应的质量限度(例如,分析化学,放行无菌检验)。例如:(1)对于批次放行,在检验报告中报告的最终含量均匀度结果被视作一个检验;(2)对于一个稳定性时间点,该时间点进行的每一个检验都被计为一个单独的检验
生产涵盖药品中所用API的涵盖设施,不要求报告稳定性OOS结果
稳定性检验中,只有支持产品实时稳定性的检验应当计入(加速稳定性检验不应计入)
如果1个批次的放行或长期稳定性检验进行了多次(例如,复检),每一次检验都应当计入
FDA意识到本指南没有讨论其它一些检验(例如,中控检验,环境检验,原辅料和包材检验)。这些检验尽管重要,但其结果不应当计入本报告
2.3 MHRA
This Medicines and Healthcare products Regulatory Agency guidance for those carrying out of specification investigations covers:
Laboratory analysis
Results
Phase Ia investigations
Phase Ib investigations
Phase II investigations
Phase III investigations
Batch disposition
2.4 EU
Delegated Regulation 2014/1252/EU
Article 7 Documentation and records
2. All quality related activities carried out during the manufacturing process shall be recorded at the time they are performed. Any deviation from the written procedures referred to in Article 7(1) shall be documented and explained. Deviations affecting the quality of the active substance or preventing the active substance from meeting the specifications referred to in Article 12(1) shall be investigated, and the investigation and its conclusions shall be documented.
6.7 实验室文件管理应当符合第4章给出的原则。实验室文件的重要部分是关于质量控制的内容,质量控制部门应当有下列文件:
iv. 结果超标和超趋势调查的规程;
6.9 某些类型的数据(如检验结果、产量、环境控制)保存方式应当便于进行趋势评价。应当处理和调查任何超趋势和超标数据。
6.35 应当超标或显著异常趋势进行调查。对任何已确认的不符合质量标准的结果或重大不良趋势,应当向相关主管当局报告。根据本GMP指南第8章的要求,应当考虑对已销售批次可能造成的影响,并征求相关官方机构的意见。
EMA 已获批基于细胞/组织的先进疗法药物的超标批次使用问答
1. What is the pathway for the exceptional administration of out-of-specification (OOS) batches of a cell/tissue based advanced therapy medicinal products (ATMPs) that have been granted a marketing authorisation?
2. Who should be notified and when?
3. How should the manufacturer/importer/MAH notify the EMA of the OOS batch(es)?
4. Are National Competent Authorities involved?
5. Are there any other obligations or expectations that the manufacturer/importer and MAH have to follow in case of an OOS batch of a cell/tissue based ATMP that has been granted a marketing authorisation?
6. What information should be provided to the patient?
EDQM Evaluation & Reporting of Results – Annex 1A: Model Template for Failure Investigation of OOS Results
The annexes of the Guideline “Evaluation and Reporting of Results”, PA/PH/OMCL (13) 113(in its current version) contain several examples of approaches to failure investigation and the re-test programme. Other approaches are possible if their scientific rationale is documented and the basic principles of the core document are followed.
The editable templates/calculation sheets are available on the EDQM Extranet in the OMCL Quality Documents section.
EDQM Evaluation & Reporting of Results – Annex 2D: Special Considerations for Animal Testing (Verification of OOS Results)
To be in line with the directive 2010/63/EU on the protection of animals used for scientific purposes, retest programmes to confirm out of specification results that involve animal tests should be designed to minimise the use and the suffering of test animals as much as possible,while on the other hand assuring that the quality of the tested medicinal product (e.g. the clinical efficacy of a tested vaccine) is appropriate for the intended use.
2.5 ICH
6.61 以下情况须有完整记录:
——超标事件的调查。
11.15 须有一定程序对任何不符合规定的检测结果进行调查、备案。这一程序须包括对数据的分析,是否存在实质性问题的评估,整改措施的分派和结论。任何对超标结果的重新抽样和(或)复检须按一套书面的程序进行。
2.6 WHO
17.12 Out-of-specification results obtained during testing of materials or products should be investigated in accordance with an approved procedure. Records should be maintained.
3. 内容集合
3.1 资源
OOS指南资料收集
 https://www.bopuyun.com/pc/subject/421
https://www.bopuyun.com/pc/subject/421OOS&OOT资料收集
https://www.bopuyun.com/pc/subject/193(OOS管理规程)
OOS管理规程)(OOS调查记录)
OOS调查记录)(无菌OOS案例)
无菌OOS案例)(OOS操作文件)
OOS操作文件)3.2 优秀文章
(OOS的处理流程)
OOS的处理流程)3.3 优秀课程
关于实验室OOS问题探讨
https://www.bopuyun.com/pc/college/76
