欢迎访问『 博普智库 』制药人必备知识工具


9013 缓释、控释和迟释制剂指导原则
 纠错
纠错
9013 缓释、控释和迟释制剂指导原则
—、概述
调释制剂,系指与普通制剂相比,通过技术手段调节药物的释放速率、释放部位或释放时间的一大类制剂。调释制剂可分为缓释、控释和迟释制剂等。其中,缓释、控释制剂与普通制剂比较,药物治疗作用更持久、毒副作用可能降低、用药次数减少,可提高患者用药依从性。迟释制剂可延迟释放药物,从而发挥肠溶、结肠定位或脉冲释放等功能。
本指导原则以口服缓释、控释和迟释制剂为重点,也可供其他给药途径的相关制剂参考。
缓释制剂,系指在规定的释放介质中,按要求缓慢地非恒速释放药物,与相应的普通制剂比较,给药频率减少一半或有所减少,且能显著增加患者用药依从性的制剂。
控释制剂,系指在规定的释放介质中,按要求缓慢地恒速释放药物,与相应的普通制剂比较,给药频率减少一半或有所减少,血药浓度比缓释制剂更加平稳,且能显著增加患者用药依从性的制剂。
迟释制剂,系指在给药后不立即释放药物的制剂,包括肠溶制剂、结肠定位制剂和脉冲制剂等。肠溶制剂,系指在规定的酸性介质(pH1.0~3.0)中不释放或几乎不释放药物,而在要求的时间内,于pH6.8磷酸盐缓冲液中大部分或全部释放药物的制剂。结肠定位制剂,系指在胃肠道上部基本不释放、在结肠内大部分或全部释放的制剂,即一定时间内在规定的酸性介质与pH6.8磷酸盐缓冲液中不释放或几乎不释放,而在要求的时间内,于pH7.5~8.0磷酸盐缓冲液中大部分或全部释放的制剂。脉冲制剂,系指不立即释放药物,而在某种条件下(如在体液中经过一定时间或一定pH值或某些酶作用下)一次或多次突然释放药物的制剂。
缓释、控释和迟释制剂的处方工艺设计可能影响其质量和疗效等,因此必须对其进行全面深入研究,并结合实际生产的具体情况,筛选出适合工业化生产的处方工艺。缓释、控释和迟释制剂体外、体内的释放行为应符合临床需求,应建立能评估体内基本情况的体外释放度实验方法和控制指标,以有效控制制剂质量,保证制剂的安全性与有效性。
二、缓释、控释和迟释制剂的制备
1. 处方工艺研究
缓释、控释和迟释制剂研发应结合临床需求与药物特性进行可行性评价,并非所有的口服药物都适合制成缓控释制剂。设计缓释、控释和迟释制剂时应考虑的因素有:药物的理化性质、生物药剂学性质、药物动力学性质、药效学性质以及临床需求等。
缓释、控释和迟释制剂的设计要依据药物的溶解性、pH对溶解度的影响、稳定性、药物的吸收部位、吸收速率、首过效应、消除半衰期、药物的最小有效浓度、最佳治疗浓度、最低毒性浓度及个体差异等,根据临床需要以及预期制剂的体内性能进行可行性评估及处方设计。药物在胃肠道不同部位的吸收特性以及制剂在肠道的滞留时间是影响口服吸收的重要因素。胃肠道不同部位的pH、表面积、膜通透性、分泌物、酶、水量等不同,在药物吸收过程中所起的作用可能有显著差异,因此,在研发前需充分了解药物在胃肠道的吸收部位或吸收窗,并在处方设计时考虑如何减小可能的个体差异。
口服缓释、控释和迟释制剂最常采用的剂型为片剂和胶囊(填充缓释小丸或颗粒),其他有缓释颗粒、缓释混悬剂等;常用的调释技术包括:膜包衣技术、骨架技术、渗透泵技术,以及胃内滞留技术、生物黏附技术、离子交换技术等。应根据药物的性质、临床用药特点、可釆用的辅料、工艺设备等情况确定具体剂型,选择合适的调释技术和体外评价方法,进行处方与工艺的筛选与优化。
在处方工艺设计和研究中,需要充分了解原料药与所用辅料的性质及彼此的相容性。由于缓释、控释和迟释制剂的制备较普通制剂更加复杂,故需对制备工艺中可能影响产品质量的环节和工艺参数进行详细的考察,确定影响制剂质量的关键工艺因素以及关键工艺参数的范围。在小试和中试生产的过程中,根据各个环节对考察参数与质量的分析结果,进一步评价缓释、控释和迟释制剂的处方工艺是否适合大生产。对多批的小试、中试和工业生产规模的产品进行质量对比研究,验证工艺的可行性与合理性,保证在设定的条件下批间差异的可控。
缓释、控释和迟释制剂的释放行为应在一定pH条件下保持稳定,符合临床需求,且不受或少受生理、饮食等因素及产品运输储存条件(温度、湿度)等各个环节的影响。良好的处方工艺及其制备过程应能保证产品的重现性和稳定性,可以通过严格的操作规程和中间控制手段,有效地解决大生产中批次间的重现性以及体内生物等效性等问题。
2. 质量控制研究
口服缓释、控释和迟释制剂的质量研究项目主要包括性状、鉴别、释放度、重(装)量差异、含量均匀度、有关物质、微生物限度、含量测定等。其中,释放度方法研究及其限度确定是口服缓释、控释和迟释制剂质量研究的重要内容。在建立释放度及其他检验项目的测定方法时,应符合《分析方法验证指导原则》(指导原则9101( 指导原则9101))。
指导原则9101))。
指导原则9101))。 应考察至少3批产品批与批之间体外药物释放度的重现性,并考察同批产品体外药物释放度的均一性。缓释、控释和迟释制剂制备工艺中若用到需要控制的有机溶剂(如包衣工艺中采用的有机溶剂),则应按《残留溶剂测定法》(通则0861(通则0861))测定,并根据测定结果及数据积累结果确定是否纳入质量标准。
通则0861))测定,并根据测定结果及数据积累结果确定是否纳入质量标准。 3. 稳定性研究
口服缓释、控释和迟释制剂的稳定性研究应按《原料药物与制剂稳定性试验指导原则》(指导原则9001(指导原则9001))进行。在稳定性考察项目方面,除一般性项目外,还应重点考察释放度的变化,分析产生变化的原因及对体内释放行为的可能影响,必要时修改、完善处方工艺。
指导原则9001))进行。在稳定性考察项目方面,除一般性项目外,还应重点考察释放度的变化,分析产生变化的原因及对体内释放行为的可能影响,必要时修改、完善处方工艺。 三、缓释、控释和迟释制剂的评价
1. 体外释放度试验
药物的体外释放行为受制剂本身因素和外界因素的影响。制剂本身因素包括主药的性质(如溶解度、晶型、粒度分布等)、制剂的处方与工艺等,外界因素包括释放度测定的仪器装置、释放介质、转速等。体外释放度试验是在模拟体内消化道条件下(如温度、介质的pH值、搅拌速率等),测定制剂的药物释放速率,并最后制订出合理的体外药物释放度标准,以监测产品的生产过程及对产品进行质量控制。结合体内外相关性研究,释放度可以在一定程度上预测产品的体内行为。对于释放度方法可靠性和限度合理性的评判,可结合体内研究数据进行综合分析。
(1)仪器装置 仪器装置的选择,应考虑具体的剂型及可能的释药机制。除另有规定外,缓释、控释和迟释制剂的体外药物释放度试验可采用溶出度测定仪进行。如采用其他特殊仪器装置,需提供充分的依据。贴剂可釆用溶出度与释放度测定法(通则0931(通则0931))测定。
通则0931))测定。 (2)温度 缓释、控释和迟释制剂的体外释放度试验应控制在37℃±0.5℃,以模拟体温;而贴剂的体外释放度试验应控制在32℃±0.5℃,以模拟表皮温度。
(3)释放介质 释放介质的选择依赖于药物的理化性质(如溶解性、稳定性、油水分配系数等)、生物药剂学性质以及吸收部位的生理环境(如胃、小肠、结肠等)。一般推荐选用水性介质,包括水、稀盐酸(0.001~0.1mol/L)或pH3~8的醋酸盐或磷酸盐缓冲液等;对难溶性药物通常不宜采用有机溶剂,可加适量的表面活性剂(如十二烷基硫酸钠等);必要时可考虑加入酶等添加物。
由于不同pH值条件下药物的溶解度、缓控释辅料的性质(如水化、溶胀、溶蚀速度等)可能不同,建议对不同pH值条件下的释放行为进行考察。
释放介质的体积一般应符合漏槽条件。
(4)取样时间点 除迟释制剂外,体外释放速率试验应能反映出受试制剂释药速率的变化特征,且能满足统计学处理的需要。释药全过程的时间不应低于给药的间隔时间,且累积释放百分率要求达到90%以上。除另有规定外,通常将释药全过程的数据作累积释放百分率-时间的释药曲线图,以制订出合理的释放度检查方法和限度。
缓释制剂从释药曲线图中至少选出3个取样时间点,第一点为开始0.5-2小时的取样时间点,用于考察药物是否有突释;第二点为中间的取样时间点,用于确定释药特性;最后的取样时间点,用于考察释药是否基本完全。控释制剂取样点不得少于5个。
迟释制剂可根据临床需求设计释放度的取样时间点。
(5)转速 缓释、控释和迟释制剂在不同转速下的释放行为可能不同,故应考察不同转速对其释放行为的影响。一般不推荐过高或过低转速。
(6)释药模型的拟合 缓释制剂的释药数据可用一级方程和Higuchi方程等拟合,即
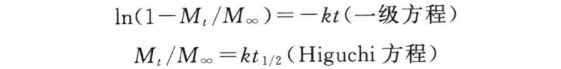
控释制剂的释药数据可用零级方程拟合,即
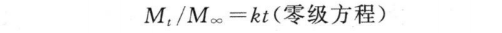
上式中,Mt为t时间的累积释放量;M∞为∞时累积释放量;Mt/M∞为t时累积释放百分率。拟合时以相关系数(r)最大而均方误差(MSE)最小为最佳拟合结果。
(7)其他 多于一个活性成分的产品,要求对每一个活性成分均按以上要求进行释放度测定。如在同一种方法下不能有效测定每个成分的释放行为,则需针对不同成分,选择建立不同的测定方法。对于不同规格的产品,可以建立相同或不同的测定方法。
2. 体内试验
对缓释、控释和迟释制剂的安全性和有效性进行评价,应通过体内的药效学和药物动力学试验。首先对缓释、控释和迟释制剂中药物特性的物理化学性质应有充分了解,包括有关同质多晶、粒子大小及其分布、溶解性、溶出速率、稳定性以及制剂可能遇到的其他生理环境极端条件下控制药物释放的变量。制剂中药物因受处方和制备工艺等因素的影响,溶解度等物理化学特性会发生变化,应测定相关条件下的溶解特性。难溶性药物的制剂处方中含有表面活性剂(如十二烷基硫酸钠)时,需要了解其对药物溶解特性的影响。
关于药物的药物动力学性质,应进行单剂量和多剂量人体药代动力学试验,以证实制剂的缓控释特征符合设计要求。推荐采用药物的普通制剂(静脉用或口服溶液,或经批准的其他普通制剂)作为参考,对比其中药物释放、吸收情况,来评价缓释、控释和迟释制剂的释放、吸收情况。设计口服缓释、控释和迟释制剂时,测定药物在肠道各段的吸收是很有意义的。食物的影响也应考虑。
药物的药效学性质应反映出在足够广泛的剂量范围内药物浓度与临床响应值(治疗效果或副作用)之间的关系。此外,应对血药浓度和临床响应值之间的平衡时间特性进行研究。如果在药物或药物的代谢物与临床响应值之间已经有很确定的关系,缓释、控释和迟释制剂的临床表现可以由血药浓度-时间关系的数据进行预测。如无法得到这些数据,则应进行临床试验和药动学-药效学试验。
缓释、控释和迟释制剂进行的生物利用度与生物等效性试验,详见指导原则9011(指导原则9011)。
指导原则9011)。 非口服的缓释、控释和迟释制剂还需对其作用部位的刺激性和(或)过敏性等进行试验。
3. 体内-体外相关性
(1)体内-体外相关性的评价方法 体内-体外相关性,指的是由制剂产生的生物学性质或由生物学性质衍生的参数(如tmax、Cmax或AUC),与同一制剂的物理化学性质(如体外释放行为)之间建立合理的定量关系。
缓释、控释和迟释制剂要求进行体内外相关性的试验,它应反映整个体外释放曲线与血药浓度-时间曲线之间的关系。只有当体内外具有相关性时,才能通过体外释放曲线预测体内情况。
体内外相关性可归纳为三种:①体外释放曲线与体内吸收曲线(即由血药浓度数据去卷积而得到的曲线)上对应的各个时间点分别相关,这种相关简称点对点相关,表明两条曲线可以重合或者通过使用时间标度重合。②应用统计矩分析原理建立体外释放的平均时间与体内平均滞留时间之间的相关。由于能产生相似的平均滞留时间可有很多不同的体内曲线,因此体内平均滞留时间不能代表体内完整的血药浓度-时间曲线。③一个释放时间点(t50%、t90%等)与一个药物动力学参数(如AUC、Cmax或tmax)之间单点相关,它只说明部分相关。
(2)本指导原则采用的方法 本指导原则中缓释、控释和迟释制剂的体内外相关性,系指体内吸收相的吸收曲线与体外释放曲线之间对应的各个时间点回归,得到直线回归方程的相关系数符合要求,即可认为具有相关性。
①体内-体外相关性的建立
(a)基于体外累积释放百分率-时间的体外释放曲线
如果缓释、控释和迟释制剂的释放行为随体外释放度试验条件(如装置的类型,介质的种类和浓度等)变化而变化,就应该另外再制备两种供试品(一种比原制剂释放更慢,另一种更快),研究影响其释放快慢的体外释放度试验条件,并按体外释放度试验的最佳条件,得到基于体外累积释放百分率-时间的体外释放曲线。
(b)基于体内吸收百分率-时间的体内吸收曲线
根据单剂量交叉试验所得血药浓度-时间曲线的数据,对体内吸收符合单室模型的药物,可获得基于体内吸收百分率-时间的体内吸收曲线,体内任一时间药物的吸收百分率(Fa)可按以下Wagner-Nelson方程计算。
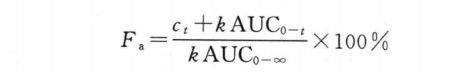
式中 ct为t时间的血药浓度;
k为由普通制剂求得的消除速率常数。
双室模型药物可用简化的Loo-Riegelman方程计算各时间点的吸收百分率。
可采用非模型依赖的反卷积法将血药浓度-时间曲线的数据换算为基于体内吸收百分率-时间的体内吸收曲线。
②体内-体外相关性检验
当药物释放为体内药物吸收的限速因素时,可利用线性最小二乘法回归原理,将同批供试品体外释放曲线和体内吸收相吸收曲线上对应的各个时间点的释放百分率和吸收百分率进行回归,得直线回归方程。
如直线的相关系数大于临界相关系数(P<0.001),可确定体内外相关。
继续阅读
在线查询结果来源于2020年版中国药典,仅供参考。由专业团队进行审核校对。

联系我们: 电话 400-805-7385 028-86051178 邮箱 contact@bopuyun.com 地址 四川省成都市高新区九兴大道6号高发大厦B幢2层
 扫码添加智库顾问
扫码添加智库顾问 扫码关注智库公众号
扫码关注智库公众号开发者: 成都博普云达科技有限公司 版本号: V24.0.11