

质量标准法规指南汇总来啦~图走起:
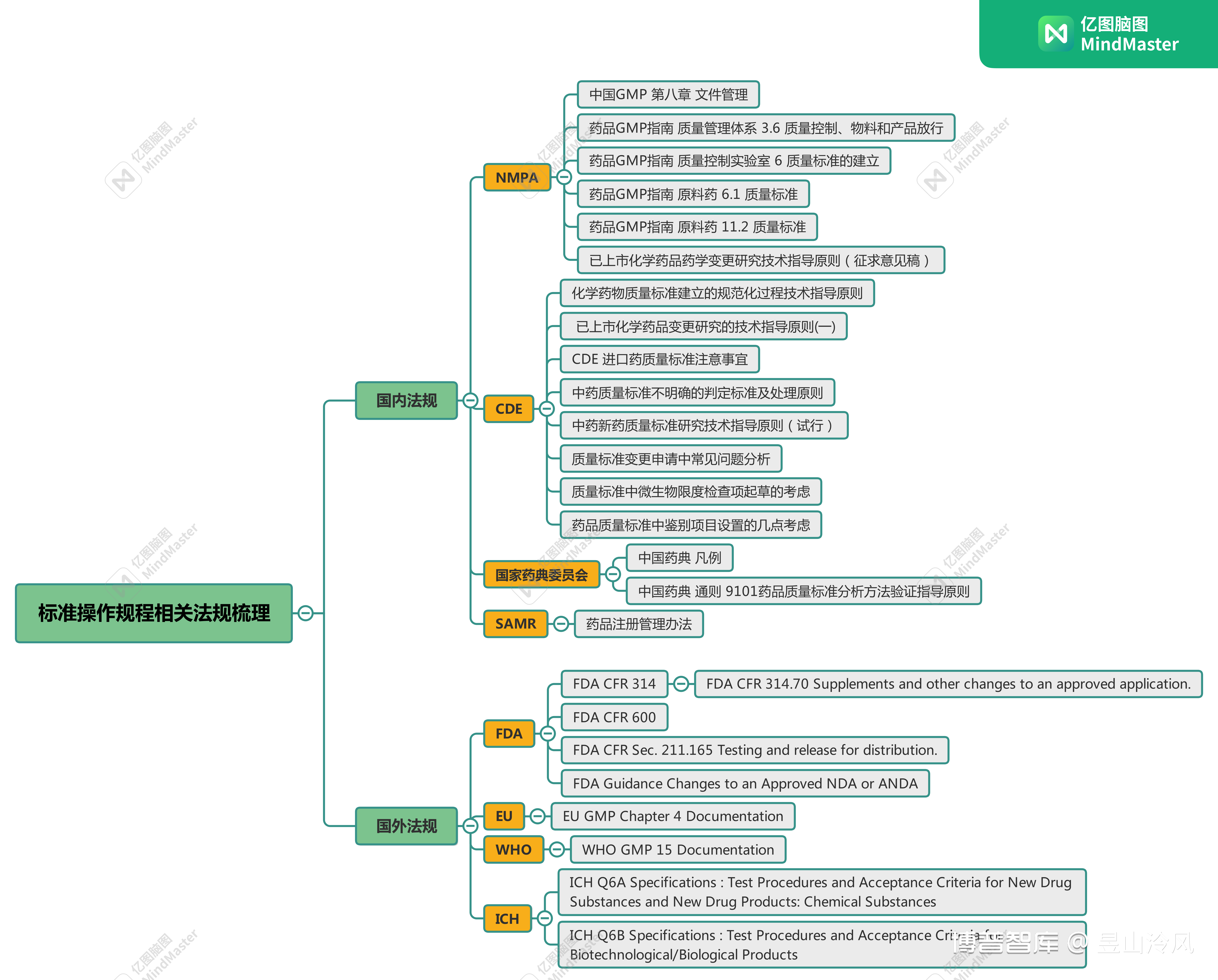
一、【基本概念】
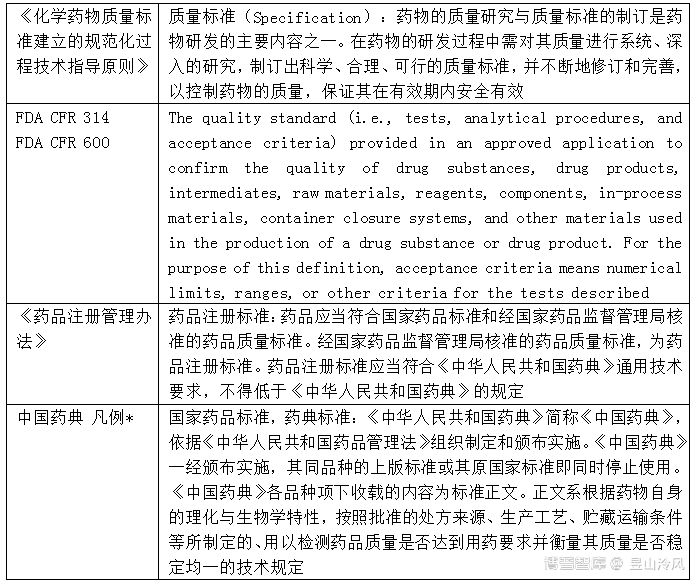
<中国药典 凡例
二、相关法规指南
【药品注册管理办法】
第八条从事药物研制和药品注册活动,应当遵守有关法律、法规、规章、标准和规范;参照相关技术指导原则,采用其他评价方法和技术的,应当证明其科学性、适用性;应当保证全过程信息真实、准确、完整和可追溯。
药品应当符合国家药品标准和经国家药品监督管理局核准的药品质量标准。经国家药品监督管理局核准的药品质量标准,为药品注册标准。药品注册标准应当符合《中华人民共和国药典》通用技术要求,不得低于《中华人民共和国药典》的规定。申报注册品种的检测项目或者指标不适用《中华人民共和国药典》的,申请人应当提供充分的支持性数据。
药品审评中心等专业技术机构,应当根据科学进展、行业发展实际和药品监督管理工作需要制定技术指导原则和程序,并向社会公布。
第三十九条综合审评结论通过的,批准药品上市,发给药品注册证书。综合审评结论不通过的,作出不予批准决定。药品注册证书载明药品批准文号、持有人、生产企业等信息。非处方药的药品注册证书还应当注明非处方药类别。
经核准的药品生产工艺、质量标准、说明书和标签作为药品注册证书的附件一并发给申请人,必要时还应当附药品上市后研究要求。上述信息纳入药品品种档案,并根据上市后变更情况及时更新。
药品批准上市后,持有人应当按照国家药品监督管理局核准的生产工艺和质量标准生产药品,并按照药品生产质量管理规范要求进行细化和实施。
第四十四条化学原料药、辅料及直接接触药品的包装材料和容器关联审评通过的或者单独审评审批通过的,药品审评中心在化学原料药、辅料及直接接触药品的包装材料和容器登记平台更新登记状态标识,向社会公示相关信息。其中,化学原料药同时发给化学原料药批准通知书及核准后的生产工艺、质量标准和标签,化学原料药批准通知书中载明登记号;不予批准的,发给化学原料药不予批准通知书。
未通过关联审评审批的,化学原料药、辅料及直接接触药品的包装材料和容器产品的登记状态维持不变,相关药品制剂申请不予批准。
【FDA CFR 314.70 Supplements and other changes to an approvedapplication.】
(6) The agency may designate a category ofchanges for the purpose of providing that, in the case of a change in suchcategory, the holder of an approved application may commence distribution ofthe drug product involved upon receipt by the agency of a supplement for thechange. These changes include, but are not limited to: (i) Addition to aspecification or changes in the methods or controls to provide increasedassurance that the drug substances or drug product will have thecharacteristics of identity, strength, quality, purity, or potency that itpurports or is represented to possess.
【FDA CFR 211.165 Testing and release for distribution】
(a) For each batch of drug product, thereshall be appropriate laboratory determination of satisfactory conformance tofinal specifications for the drug product, including the identity and strengthof each active ingredient, prior to release. Where sterility and/orpyrogentesting are conducted on specific batches of shortlived radiopharmaceuticals,such batches may be released prior to completion of sterility and/or pyrogentesting, provided such testing is completed as soon as possible.
【中国GMP 第八章文件管理】
第一百六十四条 物料和成品应当有经批准的现行质量标准;必要时,中间产品或待包装产品也应当有质量标准。
第一百六十五条 物料的质量标准一般应当包括:
(一)物料的基本信息:1.企业统一指定的物料名称和内部使用的物料代码;2.质量标准的依据;3.经批准的供应商;4.印刷包装材料的实样或样稿。
(二)取样、检验方法或相关操作规程编号;
(三)定性和定量的限度要求;
(四)贮存条件和注意事项;
(五)有效期或复验期。
第一百六十六条 外购或外销的中间产品和待包装产品应当有质量标准;如果中间产品的检验结果用于成品的质量评价,则应当制定与成品质量标准相对应的中间产品质量标准。
第一百六十七条 成品的质量标准应当包括:
(一)产品名称以及产品代码;
(二)对应的产品处方编号(如有);
(三)产品规格和包装形式;
(四)取样、检验方法或相关操作规程编号;
(五)定性和定量的限度要求;
(六)贮存条件和注意事项;
(七)有效期。
【化学药物质量标准建立的规范化过程技术指导原则】
本指导原则针对药物研发的不同情况(原料药及各种制剂)和申报的不同阶段(申请临床研究、申报生产和试行标准转正等),阐述质量研究和质量标准制订的一般原则和内容,重点强调药物研发的自身规律、质量研究和质量标准的阶段性,以及质量标准建立的规范化过程。
本指导原则旨在引导研发者根据所研制药物的特点和药物研发的自身规律,理清研究思路,规范质量研究、质量标准的制订,以及质量标准的修订和完善的过程,提高质量标准的质量。
本指导原则的基本内容共分四个部分:质量标准建立的基本过程、药物的质量研究、质量标准的制订和质量标准的修订。
本指导原则适用于化学药,包括新药、进口药和已有国家标准的药品。
EU GMP Chapter 4 Documentation
WHO GMP 15 Documentation
FDA 21 CFR Sec. 211.110(a) Sampling andtesting of in-process materials and drug products.
FDA 21 CFR Sec. 211.160 Generalrequirements.
FDA 21 CFR Sec. 211.166 Stability testing.
FDA CFR 211 Sec. 211.192 Production recordreview
......
【 FDA Guidance Changes to an Approved NDA or ANDA】
Specifications (i.e., tests, analyticalprocedures, and acceptance criteria) are the quality standards provided in anapproved application to confirm the quality of drug substances, drug products,intermediates, raw materials, reagents, components, in-process materials,container closure systems, and other materials used in the production of a drugsubstance or drug product. For the purpose of defining specifications,acceptance criteria are numerical limits, ranges, or other criteria for thetests described.
This guidance list the examples of MajorChanges (Prior Approval Supplement), Moderate Changes (Supplement - ChangesBeing Effected), Minor Changes (Annual Report)
三、执行
1.药物的质量研究是质量标准制订的基础,质量研究的内容应尽可能全面,既要考虑一般性要求,又要有针对性。确定质量研究的内容,应根据所研制产品的特性(原料药或制剂),釆用的制备工艺,并结合稳定性研究结果,以使质量研究的内容能充分地反映产品的特性及质量变化的情况。参考CDE 化学药物质量标准建立的规范化过程技术指导原则,药品GMP指南质量管理体系 3.6 质量控制、物料和产品放行,药品GMP指南质量控制实验室 6 质量标准的建立,ICH Q6A Specifications : Test Procedures and Acceptance Criteria forNew Drug Substances and New Drug Products: Chemical Substances,ICH Q6BSpecifications : Test Procedures and Acceptance Criteria forBiotechnological/Biological Products.
2.药品研制和生产各环节是紧密关联的,生产工艺、处方中已有药用要求的辅料、质量标准等某一方面变更可能对药品安全性、有效性和质量可控性带来全面的影响。参考CDE 已上市化学药品变更研究的技术指导原则(一).
药品处方、生产工艺、场地、批量、质量标准等某一个方面的变更可能对药品安全性、有效性和质量可控性带来全面的影响。药品某一项变更往往不是独立发生的。例如,批量变更往往同时伴随生产设备及生产工艺的变更,处方变更可能伴随或引发药品质量标准变更,增加规格可能会调整处方等。原辅包的各项变更,如生产工艺的变更、生产场地的变更、批量的变更、质量标准的变更等,可能对原辅包的质量存在影响,进而对制剂产生影响。参考NMPA 已上市化学药品药学变更研究技术指导原则(征求意见稿).
3.对原料、中间体、原料药和标签及包装材料应建立适当的质量标准。原料药应符合药典标准。起始物料/原料/中间体不必须执行药典标准,而应按照产品需求建立适当的标准。原料质量标准的建立可以参照研发报告的相关内容,原料采购合同可以引入相关质量标准。接收、取样、检测部门应熟悉相关的标准。对过程控制应建立适用的标准。参考药品GMP指南原料药 6.1 质量标准、药品GMP指南原料药 11.2 质量标准
4.药物质量标准的建立主要包括以下过程:确定质量研究的内容、进行方法学研究、确定质量标准的项目及限度、制订及修订质量标准。可以参考化学药物质量标准建立的规范化过程技术指导原则和ICH Q6A。
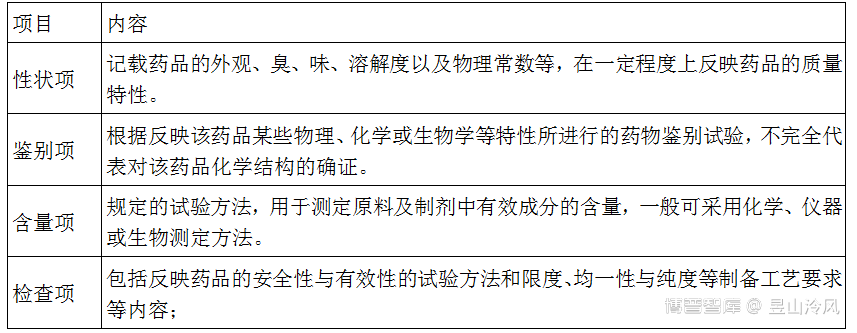
在建立质量标准的过程中,应考虑质量标准的适用性。包括哪些项目放入质量标准,哪些不放入质量标准,质量标准限度的适宜性。
质量标准建立的项目:如果原料药没有收入药典标准,还可以参考ICH等行业指南的要求,如果是仿制药,可以参考原研药或RLD或已上市药品的质量研究,还应该考虑原料药的制造工艺、对应制剂的制造工艺和剂型要求、产品性质研究、杂质研究、稳定性数据、安全性数据和批分析数据等。
对于质量标准中不建立的项目,例如某些一类溶剂残留的检测,一方面应该在原辅料中进行一类溶剂含量的控制,另一方面对原料药成品进行验证性实验,证明在工艺控制下,成品中不含这种一类溶剂,因此可以不做放行检测。再例如有些潜在的杂质(中间体)不会出现在API成品中,应该在杂质确认的时候列出,但是在质量标准建立的时候,提供证据证明工艺可以除去这些杂质,并且使用经过验证的检测方法对成品进行检验后没有发现这些杂质,就可以不在质量标准中列出这些潜在杂质的检测。
对于质量标准限度,应考虑检测方法的误差、工艺的波动和生产部门的实际操作 , 确定适宜的限度,这里应该参考批分析数据 , 如果限度放得太宽,即使出现了偏差也无法发现 , 可能就不符合GMP要求的持续稳定可控的生产工艺。
在实际操作中 , 如果是自己制剂配套的API,会根据制剂的标准再加上ICH的要求进行一下紧缩。如果是外部客户 , 一般客户会提出要求。总的来说 , API的标准时建立在制剂的要求上的。
四、其他资料:
【生物制品的质量标准相关资料】变态反应原(变应原)制品质量控制技术指导原则人基因治疗研究和制剂质量控制技术指导原则人体细胞治疗研究和制剂质量控制技术指导原则人用单克隆抗体质量控制技术指导原则细胞培养用牛血清生产和质量控制技术指导原则人用重组DNA制品质量控制技术指导原则重组制品生产用哺乳动物细胞质量控制技术评价一般原则
【CDE关于质量标准的电子刊物】进口药质量标准注意事宜质量标准中微生物限度检查项起草的考虑药品质量标准中鉴别项目设置的几点考虑药品注射剂质量标准中安全性检查的几点思考制订抗生素有关物质标准的指导原则(草案)-欧洲药物管理局(EMA)质量标准变更申请中常见问题分析撰写质量标准中无菌检查项内容需注意的几个方面准备进口化学药品质量研究和质量标准资料的几点建议

