

September 2, 2021
R.D. McDowall
Spectroscopy, September 2021, Volume 36, Issue 9
Pages: 15–22, 58
Columns | Column: Focus on Quality
美国食品和药物管理局(FDA)对计算机化系统验证(CSV)有一个新的方法,称为计算机系统保证(CSA)。在没有发布指南草案的情况下,我们是否进入了一个通过介绍和公布来监管的时代?因此,CSA是否有变成 "完全愚蠢的保证 "的风险?
计算机化系统验证(CSV)有一个不令人振奋的名声,即它是一个缓慢的、没有附加值的活动,只会浪费时间和推迟新软件的实施。这是一个准确的描述吗?
作为一个与CSV打交道超过35年的人,我想说这取决于。这里有两个CSV的例子,一个是使用CSV是崇高的,另一个是使用CSV是可笑的。
- 崇高的例子。绘制并改进你的流程,你的预期使用需求就是从这些流程中写出来的。然后,应用有效的风险管理和科学合理的逻辑,利用供应商开发和应用配置来集中测试。为什么不测试一个功能的理由和为什么要测试另一个功能的理由一样重要。这种方法是管理合规成本与不合规成本的一个例子,在最近的 "关注质量 "专栏中讨论过(1)。
- 荒谬的例子。一个组织对CSV有一个一刀切的方法。当企业的CSV标准操作程序(SOP)规定你必须写三份规范文件(用户需求规范、功能规范和设计规范),但紫外光谱仪的预期用途是测量一到两个波长的样品吸光度时,你就知道你在浪费精力了。一个详细的风险评估为无附加价值的火苗添了一把火,随后又要求提供详细的测试说明,并附上大量的屏幕截图,以证明每个测试的每个步骤都已执行。
一刀切的验证方法缺乏灵活性,不能根据预期的用途来定制每个验证,这就注定了任何受监管的实验室都要面对堆积如山的纸张。你可以看到为什么CSV会有不好的名声;CSV不是应用常识来产生系统的商业利益,而是一种不灵活的方法,再加上制药业的极端保守性,使其成为一个老式的、过时的过程。你应该对供应商的开发和测试、应用软件类别以及系统创建的记录的影响有一个准确的评估,以便将CSV工作集中在最需要的地方。灵活性是游戏的名称。
CDRH:质量的案例
大约10年前,FDA的设备和放射健康中心(CDRH)开始了 "质量案例 "倡议,旨在审查医疗器械公司在监管合规方面的问题。到2015年,确定的领域之一是CSV,原因如下。
- CSV没有展示它的预期用途,而是旨在制作文件,让审计师和检查员保持沉默。
- CSV是延迟的代名词,因此,它最终成为一种必要的罪恶,而不是一种增值活动。
- 风险评估是复杂的,繁琐的,昂贵的。
- 80%的测试问题是由于测试人员或测试说明中的错误造成的。
- 行业普遍以监管负担为借口,不利用技术进步来改善CSV。
因此,FDA成立了一个CDRH和行业联合小组,为医疗器械行业使用的计算机系统制定新的验证方法,目的是遵循负担最小的方法,称为计算机系统保证(CSA)。
负担最小的方法
2002年发布的《软件验证的一般原则》是CDRH对行业的指导。在第2.3节中,它指出。
我们相信,在医疗器械监管的所有领域,我们都应该考虑采用负担最小的方法。本指南反映了我们对相关科学和法律要求的仔细审查,以及我们认为对你们来说,符合这些要求的负担最小的方法(2)。
本节最后邀请,如果公司知道有更好的方法来验证软件,请与该机构讨论。该指南在关于验证范围的第4.8节中做了进一步的阐述,我们逐字逐句地引用。
验证范围应基于软件的复杂性和安全风险,而不是基于公司规模或资源限制。验证活动、任务和工作项目的选择应与软件设计的复杂程度以及将软件用于指定用途的风险相称。对于低风险的设备,可以只进行基线验证活动。随着风险的增加,应增加额外的验证活动以覆盖额外的风险。验证文件应足以证明所有的软件验证计划和程序都已成功完成(2)。
这里的关键启示是要关注预期用途、风险和所使用软件的性质。有20年历史的FDA指南发出了一个明确的信息,套用一句话说,重要的是不要用合规性杀死自己。长期以来,制药业没有从一个规避风险的行业发展到一个风险管理的行业。
FDA:CDRH和CDER中心
FDA分为几个中心。有两个中心在本专栏中被详细讨论。
- 药品评估和研究中心(CDER)。该中心负责制药业。适用的《药品生产质量管理规范》(GMP)法规是21 CFR 211,该法规于1978年首次发布,并在2008年进行了小幅更新(3,4)。由于这些法规的年代久远,没有明确要求对计算机系统进行验证。
- 设备和放射卫生中心(CDRH)。这个中心负责医疗设备,包括扫描仪。CDRH使用的GMP法规是21 CFR 820 (5),是基于1990年代的ISO 13485 (6)。GMP对验证医疗设备中使用的软件(21 CFR 820.30)和用于过程控制和质量管理系统(QMS)的软件(21 CFR 820.70[i])有具体要求(5)。
用于控制或操作医疗设备的软件在你购买时已经得到验证,这与制药业使用的软件形成鲜明对比,后者在你购买时没有得到验证(尽管许多供应商希望你相信它得到验证),实验室必须进行CSV,以证明它适合预期用途,这是基于业务需求和自动化过程。请记住,CSA主要针对的是医疗器械,而不是制药业。
CSA原则和试点项目
行业-CDRH联合小组制定的CSA原则为:。
- 专注于某项应用的预期用途,通常是医疗设备
- 从记录到批判性思维的转变
- 允许无记录的测试
- 只使用值得信赖的供应商
- 证据应该为测试过程增加价值,也就是减少软件错误的数量,以及证明预期用途
- 使用自动化来帮助流程,如需求管理和测试
试点项目被用来验证和完善CSA方法。自2017年以来,FDA工作人员和各个试点项目的成员都有一些演讲和出版物。到目前为止,情况良好。
然而,尽管CSA的行业指南草案自2018年以来一直在CDRH的文件清单上,但该局没有出现任何文件,这是一个问题。
等待戈多?
CSA指南的悬而未决,相当于监管部门的 "等待戈多"(译者注,一个经典的悲剧舞台剧,一个“什么也没有发生,谁也没有来,谁也没有去’’的悲剧。剧中的两个主要人物在舞台上吐出了两个多小时的垃圾,而戈多却从未出现过。)。
对比一下美国和欧洲在法规方面的不同做法是很有意思的。自2011年以来,欧盟(EU)的GMP已经更新了第一部分九章中的八章,包括几个附件,如11和15。事实上,第4章和附件11正在被再次修订,以加强数据完整性原则。相比之下,1978年发布的美国GMP(21 CFR 211)只在2008年更新过一次,增加了一个影响生产的条款:211.68(c) (4)。
我认为,美国食品和药物管理局,特别是CDRH,没有发布关于CSA的指导草案,是无能和不专业的。
FDA不更新法规,而是以一级行业指导文件或在FDA网站上公布的二级问题和答案部分来发布建议。让我们把重点放在一级指南上,这些指南通常以草案的形式发布,供行业评论,经过长时间的思考后,最终版本才会发布:
1. Part 11的范围和应用指南,于2003年2月发布,并于同年8月定稿(7)。(相对来说,快速
发布指南的例子了)
2. 数据完整性和遵守cGMP指南的进展较为缓慢,从2016年4月的草案到2018年12月的最终版本(8)。
3. 规格外指导草案于1998年9月完成,但最终版本直到2006年10月才发布(9)。
4. GMP方法验证指南草案于2000年发布。最终指南于2015年7月发布,距离草案仅有15年时间。(10).
可以说,所有的指导文件都在每一页上印有 "包含无约束力的建议 "的字样,意味着内容可能难以执行。
逃出魔瓶的精灵
在没有行业指南草案的情况下,有CDRH官员和试点项目的行业成员的发言,也有文章、白皮书和行业指南的发表。现在的情况是,我们把行业解释放在了监管马之前。通常情况下,情况恰恰相反。美国食品和药物管理局发布指南草案供行业评论,然后在警告信中引用后,由行业实施演讲和出版物。这次不是,因为精灵已经出了瓶子。休斯顿,我们有一个更大的问题(译者注,此处用梗NASA经典台词:Houston, we have a bigger problem)。
通过介绍和公布进行监管
法规和一级法规指导文件必须经过适当的程序。这个过程包括发布草案供行业评论,适当的时候进行修改,然后是最终版本。对于法规来说,在《联邦公报》上发表的最终版本包含了行业意见的摘要,以及机构的审查和回应,机构要么拒绝这些意见,要么采取行动。你可以在1997年3月的《联邦公报》上看到21 CFR 11的情况。该法规有3页,序言评论有35页(11)。
然而,对于CSA,情况是不同的。美国食品和药物管理局列出了他们每年将发布的指导文件,其中关于CSA的指导文件至少已经在名单上三年了。觊觎并不是该局不采取行动的借口,因为在大流行之前就已经承诺了该指南,而且在家工作应该能够发布指南。相反,概述如何进行CSA的演讲和出版物正像垃圾一样被扔掉。然而,必须注意的是。
- 没有遵循适当的程序。
- 这是通过介绍和出版来监管。
- 美国食品和药物管理局已经放弃了它的监管作用。
没有行业指导草案,我们无法看到FDA对CSA的目标是否被过滤、加强或颠覆。换句话说,人们对所提出的观点的监管完整性感到担忧。作为一个受监管的行业,我们不能仅仅根据传言来改变方向。我们需要一份指南草案。但是...
我们需要CSA吗?
围绕CSA的所有问题,出现了一个重要问题。我们到底是否需要它?我们现在是否已经有了针对制药业的法规和指南,使我们能够灵活地做CSA指南中声称的事情?在我看来,答案是肯定的,我将在下面的章节中解释。当然,这是我的解释,但由于没有针对行业的指导,我的讨论可能存在漏洞。
监管的灵活性
下面是 "软件验证的一般原则 "(General Principles of Software Validation)中的两段话。第2.1节简单地指出。
本文件以公认的软件验证原则为基础,因此可以适用于任何软件(2)。
第2节中概述的指导范围指出:
该指南建议整合软件生命周期管理和风险管理活动。根据预期的用途和与要开发的软件相关的安全风险,软件开发者应确定具体的方法、要使用的技术组合,以及要应用的努力程度"(2)。
欧盟GMP附件11和15中反映了对所有软件的灵活的基于风险的CSV方法。附件11的第1条着重于风险管理。
风险管理应贯穿计算机化系统的整个生命周期,并考虑到病人安全、数据完整性和产品质量。作为风险管理系统的一部分,关于验证和数据完整性控制的程度的决定应基于对计算机化系统的合理和记录的风险评估(12)。
该条例明确指出,验证和数据完整性控制的程度应基于系统对产品和病人所造成的风险。实验室的产品可以解释为提交的数据或病人的医药产品。第1条隐含的意思是,一刀切的验证方法是不合适的。重要的是要使验证适合于系统,而不是相反。
Chris Burgess和我本人于2013年在本专栏发表了分析仪器和系统的系统级风险评估(13)。这种方法可以用来将每个人归入最新的USP <1058>组B和C子类型(14)
,以确定所需的资格和验证程度。接下来,我们有关于资格和验证条款2.5的附件15。
在适当的情况下,资质文件可以合并在一起,例如,安装资质(IQ)和运行资质(OQ)(15)。
这是有道理的,当工程师安装和鉴定你的下一个光谱仪时,
你可以有一个单一的安装鉴定(IQ)或操作鉴定(OQ)文件,将这两个活动结合到一个文件中。一份用于执行前审查和执行后批准的文件很有吸引力。这种方法在USP <1058>中得到了反映,它允许在适当的情况下将资格认证活动和相关文件合并在一起(IQ和OQ)(14)。但为什么要止步于此?
一个综合的验证文件
还记得我们在介绍中讨论过的紫外光谱仪吗,该系统所做的就是测量几个波长的吸光度?为什么不把附件15第2.5条作为一个合乎逻辑的结论,把所有的验证要素合二为一?该文件应该包括。
- 只有预期的使用要求(即工作范围、精度、线性度、预期性能和符合性)。
- 通过测试参考和系统使用的SOP链接,文件内的需求追溯性很容易建立。
- 要么交叉引用供应商的安装和调试文件,如果没有,可以在综合文件中写上同样的内容。
- 测试准备和测试方法的要求,及如何限制对记录的任何造假、排除在外等。
- 测试仪器识别
- 测试案例以证明系统符合预期的要求
- 测试执行过程,记录并解决任何测试问题
- 审核总结报告和放行声明
这听起来是一个很长的清单,但由于只关注预期使用要求,这可能是一个相对较短的文件。过程的控制将通过SOP或验证总计划进行。我已经为基于GAMP软件第3类和简单的第4类的系统实践并发表了这样的方法,即使产生的数据被用于批量发布或提交给监管机构(16)。关键是附件11(12)第1条所要求的文件化的风险管理。
我是否需要进行风险评估?
我知道你在想什么,《软件验证的一般原则》(2)建议用风险评估来集中工作,欧盟GMP附件11说你的需求风险管理应该在计算机化系统的整个生命周期内应用(12)。然而,这并不意味着你必须总是进行GAMP 5(17)中描述的定性的故障模式效果分析(FMEA)。
让我给你举个例子,说明为补救数据完整性审计发现而进行的风险评估的愚蠢性。所有用户共享同一个用户账户。猛吸一口气:没有行动的归属!接下来发生了什么?实验室随后用FMEA风险评估法评估了共享用户账户的风险和影响,并对所有要素进行了数字评估。在评估结束时,会产生一个单一的数字,并与一个尺度进行比较,以确定它是关键的、主要的、次要的还是低的。不出所料,这个数字表明这是一个关键问题(与审计师的发现相同!)--现在才触发补救措施。猜猜看,决议是什么?是的,给每个用户提供他们自己的账户。负担最小的方法?我不这么认为。
这有多愚蠢呢?它是死于合规性。一旦在审计中被发现,你就知道你有一个关键的漏洞。你已经不符合规定了。修复它。当你知道唯一可能的结果是什么时,就不要进行风险评估了。只要修复它。附件11并不要求进行风险评估;它要求应用风险管理。审计已经确定了风险;补救措施是给每个用户提供他们自己的账户,这样做更快、更容易,而且符合GMP要求。正如退休的挪威GMP检查员Audny Stenbråten所说,"使用风险评估并不为不符合规定提供借口"。O'Donnell等人有一篇有趣的文章,题为 "质量风险管理中的形式主义概念",建议进一步阅读这个话题(18)。
与其仅仅应用GAMP指南(17)中描述的单一风险评估方法,不如应用一些方法,对应用软件和IT基础设施实施可扩展的风险评估方法(19)。
充分利用供应商的发展
CSA的倡导者提到可信的软件供应商。让我们回到2008年,GAMP 5的出版,FDA经常引用,它在2.1.5、7和8.3节以及附录M2中讨论了利用供应商的参与来进行供应商评估(17)。其中有关于利用供应商测试来验证的评论。为了利用供应商测试并减少验证工作,你必须做更多的工作,而不仅仅是发出一份问卷让供应商填写,让QA把问卷放在档案柜或文件管理系统中。这个过程需要一个主动的评估,审查第4类软件应用的软件开发程序和实践,如:。
- 什么是软件开发的生命周期?
- 使用什么软件开发工具?
- 如何规定和理解对系统的要求?
- 软件代码审查的广泛性和准确性如何,它们是否得到了落实?
- 如何管理软件的构建和配置?
- 回归测试是否系统地进行并准确地报告?
- 正式测试的范围有多大?
- 如何识别、分类和解决软件错误?
- 什么是放行过程?
在了解供应商的质量管理体系和软件开发方面的投资越大,你就越能依赖供应商的决策和流程。这种类型的评估不适合做问卷调查,而是现场或远程审计。它至少需要一天的时间来执行。你正在寻找一个强大的软件开发过程。确定两到三个需求,并通过供应商的开发过程追踪它们。这些工作的范围有多大,这是否让你对供应商有信心?作为评估的一部分,包括供应商必须回答的关于合作和分享仪器和软件问题及更新信息的问题。你想要一个你可以信任的供应商。这个评估必须记录在报告中,因为它是你利用供应商的发展进入你的验证项目以减少工作量的基础。
这些信息可按以下方式使用。
- 被评估的应用程序是一个可配置的软件产品(GAMP类别4)。
- 虽然在应用层面被归类为第4类,但有许多属于第3类的功能只是简单的参数化,例如选择一个波长来测量样品的吸光度。
- 评估URS中的要求,并与应用进行比较,然后将每个要求归为第4类(已配置)或第3类(参数化)。
- 如果评估报告是肯定的,所有第3类要求都被认为是由供应商验证的,并在用户验收测试阶段进行隐性测试(20)。
在这里投入少量时间就可以减少任何系统的用户验收测试的数量和程度。这意味着不需要CSA,因为法规和行业指导已经建议采用这种方法超过10年。
非脚本测试
据称CSA的方法之一是非脚本测试,但在没有FDA指南草案的情况下,需要谨慎对待。我会提醒任何受监管的制药实验室说他们做了无记录的测试,特别是在今天的数据完整性环境中。请记住,控制医疗设备的软件是根据21 CFR 820进行验证的,不能配置,所以有一种解释是,非脚本测试可以在测试期间进行,目的是发现错误,而不是在正式的发布测试中。
如何将其应用于制药实验室中使用的软件?
其中一个领域是原型开发,以学习如何配置和使用一个应用程序。只要这个阶段在验证计划中有所描述,无文件的原型开发是可以接受的,其交付物是包含商定的软件设置的应用配置规范。
批判性思维(Critical Thinking)
批判性思维不是不顾一切的测试方法,而是应该着重于证明系统的预期用途和相关的合规功能,以支持产品开发或发布。
- 考虑一个简单的密码过期时间设置为90天的情况:除了确认过期时间已经在应用程序中配置,你现在要做什么?
- 我审计的一个系统有一个测试脚本,从11月开始,到第二年2月才结束。为什么这么长时间才运行脚本?答案是,我们在等待密码过期!"。
- 另一种方法是将密码有效期重置为一天,以测试它是否过期,然后重置为90天。在目前的数据完整性环境下,这并不是证明你可以轻易改变配置设置的最好方法。
- 如何思考这个问题:如何衡量密码的有效期?它使用一个同步到网络时间服务器的系统时钟,而网络时间服务器本身也同步到一个可信的时间源。该时钟是一个已知频率的振动石英晶体,计算机对其进行计数,然后将其转换为时间。你所做的只是检查计算机是否能够计数。你为什么要测试这个?
- 另外,如果密码过期失败,90天后密码仍然有效,那么风险是什么?答案是,风险是最小的。如果在使用过程中过期失效,它是很容易被发现的。此外,允许密码到91天后才被发现,对产品的风险几乎没有影响。角色和密码仍然在位,并能发挥作用。
测试假设、排除和限制
Barry Boehm在1970年为美国军方撰写的一份报告中解释说,不可能对软件进行详尽的测试(21)。我们在计算机的日常使用中看到了这一点,安全更新、补丁、小版本、快速修复,或任何用于修复错误的名称。然而,我们的重点是测试效率,以及如何将重点放在证明系统的预期用途的重要内容上。除了利用供应商开发外,减少测试工作量的关键是记录你在测试方法中的假设、排除和限制。仅仅因为你有一个需求并不意味着你必须盲目地测试它:你需要客观地思考。
例如,如果一个应用程序有100种不同的访问权限,而你要五个不同的用户角色,这就导致了500种不同的组合。希望你不会测试所有的组合,但你会测试多少种?你将如何论证你的方法?这就是文件化的假设、排除和你的测试方法的限制的作用,它记录了什么、如何、以及你的测试范围的任何理由,以及你如何利用供应商的发展。如果你要从测试中排除特定的用户需求,说明你为什么要这样做(20)。
硬币的另一面是在系统的统一注册系统(URS)中包括额外的要求,以备将来可能被使用。如果进行测试,这些要求可能会导致额外的工作,如果它们从未被使用,则价值为零。例如,如果一个系统用于定量分析,你是验证所有的校准模式还是只验证现在使用的那些?更好的方法是在最初的验证中专注于当前的要求。如果以后需要,可以评估其他的软件功能,但不用于监管工作。如果你想使用它们,提出一个变更请求,在更新的URS中包括这些要求,验证它们是否按预期工作。对于独立的光谱仪系统,你不太可能有单独的测试实例来评估它们,所以这是在验证过的系统中添加新功能的唯一实用方法。
测试说明
CSV存在的祸根是测试文档:你将在什么水平上记录细节?你是用受过训练的用户还是从街上拖一个人去测试你的软件?如果是前者,与后者相比,你可以减少所需的细节。不要把测试人员当作天真无邪的人,用麻木的详细说明来对待他们,测试人员是受过教育和训练的;把他们当作成年人来对待。
表一比较了规避风险的用户和受过训练的用户的测试指令。一般来说,对于左栏所示的规避风险的指令,每条指令都需要记录下观察到的结果,注明日期,并签名。如果你真的很不走运,你会在每个步骤都有一张截图。相比之下,更好的方法是给受过训练的用户一个更简单的指令,如表一右栏所示。训练有素的用户会知道如何稳定地执行这个指令。请注意,测试指令的质量取决于测试人员和测试人员对软件的了解。对系统的培训和经验越多,就越容易写出更简单的指令和执行 em。
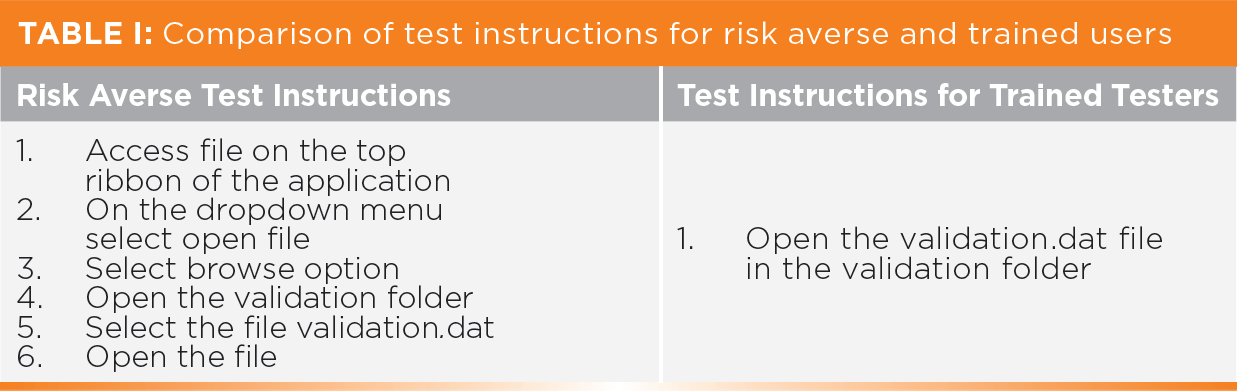
不要在每个测试步骤上注明日期和首字母,只需让测试者和审查者在每页的底部签名和注明日期,就像你在实验室笔记本上做的那样。此外,一些测试说明可能会指示测试者进入到应用程序的不同功能。如果是这样,为什么测试需要对这种指示的预期和观察结果?
你能在减少测试文件方面走得更远吗?当然,你可以这样做,但在没有指导草案的情况下,你为什么敢这样做?
无止境的截图(Screenshots at Dawn)
截图是CSV的祸根:它们被过度使用,在大多数情况下,其价值为零。如果用于记录测试中的每一步,就表明了对计算机验证的过度谨慎和规避风险的方法,也是对执行、整理、审查和保留所需资源的绝对浪费。如果少用的话,截图可以增加文件的价值,在没有其他方法记录的情况下,屏幕上的瞬时信息。GAMP的《测试良好实践指南》强调了这一点,即只有在有价值的情况下才进行屏幕截图。
然而,如果屏幕上的瞬时按摩也会导致审计跟踪条目,为什么要截图?使用审计跟踪条目来自动记录活动。通过这种方式,你不仅可以通过测试分析功能来节省时间,还可以同时验证审计跟踪功能。这样一来,既增加了测试的优雅性,又减少了测试的时间。
另一种记录测试的方法是利用屏幕视频来记录所有正在进行的工作。如果在测试计划和测试指导大纲中描述得当,利用屏幕视频是记录证据的完美方式。评审员可以随机选择段落进行评审(22)。
测试的自动化
测试自动化是非常好的,但它也有注意事项。测试自动化的最佳时机之一是在软件开发中,如回归测试,以查看新软件构建中的现有功能是否仍按预期工作。因此,测试自动化是快速的,如果一个新的构建没有通过回归测试,那么就不会进行人工测试,直到错误被修复。人工测试往往集中在开发中的版本中增加的新功能。
在验证实验室系统中使用自动化测试,最好集中在网络化的应用上,因为大多数自动化测试工具,如HP应用生命周期管理(ALM),都是网络化的。然而,测试工具也有与手工测试相同的问题,即你需要知道你所测试的应用软件和测试步骤的详细程度。Frewster和Graham指出,当第一次使用自动化测试工具时,编写测试套件的时间比手工测试要长。此外,需要10次以上的执行才能使测试工具的投资得到回报(23)。自动化测试工具的优点是通过登录凭证自动归因于行动,同时执行,并能够轻松、快速、自动地捕获和整合电子文档证据(包括可怕的屏幕截图)。这使得审查更加快速和容易。
当你试图考虑在独立系统上进行自动化用户验收测试时,现实会咬住不放,因为这更有问题。你将如何把测试工具加载到光谱仪系统上?你将如何管理记录的证据并在验证后安全地储存它?别告诉我你会用U盘(24)!你会用什么?
摘要
尽管FDA已经制定了CSA,但由于无法发布有关该主题的指导草案,导致通过介绍和公布进行监管。缺乏指导草案并不是一个适当的监管过程。然而,是否需要CSA?在目前的法规和行业指导中,有足够的现有灵活性,而制药业并没有利用。如果制药业阅读法规,将CSV理解为一种商业利益和投资保护,而不是监管的开销,这将使CSV过程更加简单和容易。然而,FDA的无能是为了让顾问们有收益吗?
鸣谢
我想感谢Chris Burgess、Mark Newton、Yves Samson、Siegfried Schmitt和Paul Smith在撰写本专栏时提出的有益意见和建议。
参考文献 References
1.
R.D. McDowall, Spectroscopy 35(11), 13–22 (2020).
2.
US Food and Drug Administration, Guidance for Industry General Principles of Software Validation (FDA, Rockville, Maryland, 2002).
3.
Part 211 - Current Good Manufacturing Practice for Finished Pharmaceuticals. Federal Register 43(190), 45014–45089 (1978).
4.
21 Code of Federal Regulations (CFR), 211 Current Good Manufacturing Practice for Finished Pharmaceutical Products (Food and Drug Administration, Silver Spring, Maryland, 2008).
5.
21 Code of Federal Regulations (CFR), 820 Quality System Regulation for Medical Devices (Food and Drug Administration: Rockville, Maryland, 1996).
6.
ISO 13485: Medical devices−Quality management systems−Requirements for regulatory purposes (International Standards Organization: Geneva, Switzerland, 2016).
7.
US Food and Drug Administration, FDA Guidance for Industry, Part 11 Scope and Application (FDA, Rockville, Maryland, 2003).
8.
US Food and Drug Administration, FDA Guidance for Industry Data Integrity and Compliance With Drug CGMP Questions and Answers (FDA, Silver Spring, Maryland, 2018).
9.
US Food and Drug Administration, FDA Guidance for Industry Out of Specification Results (FDA, Rockville, Maryland, 2006).
10.
US Food and Drug Administration, FDA Guidance for Industry: Analytical Procedures and Methods Validation for Drugs and Biologics (FDA, Silver Springs, Maryland, 2015).
11.
21 Code of Federal Regulations (CFR), Part 11, Electronic records; electronic signatures, final rule, in Title 21 (Food and Drug Administration: Washington, D.C., 1997).
12.
European Commission Health and Consumers Directorate-General, EudraLex: Volume 4 Good Manufacturing Practice (GMP) Guidelines, Annex 11: Computerized Systems (European Commission, Brussels, Belgium, 2011).
13.
C. Burgess and R.D. McDowall, Spectroscopy 28(11), 21–26 (2013).
14.
General Chapter <1058> “Analytical Instrument Qualification,” in United States Pharmacopoeia (United States Pharmacopoeia Convention, Rockville, Maryland).
15.
European Commission Health and Consumers Directorate-General, EudraLex: Volume 4, Good Manufacturing Practice (GMP) Guidelines. Annex 15: Qualification and Validation (European Commission, Brussels, Belgium, 2015).
16.
R.D.McDowall, Quality Assurance Journal 12, 64–78 (2009).
17.
ISPE, Good Automated Manufacturing Practice (GAMP) Guide, version 5 (International Society for Pharmaceutical Engineering, Tampa, Florida, 2008).
18.
K. O’Donnell, D. Tobin, S. Butler, G. Haddad, and D. Kelleher, Understanding the Concept of Formality in Quality Risk Management (IVT Network, 2020).
19.
R.D.McDowall, Quality Assurance Journal 9, 196–227 (2005).
20.
R.D.McDowall, Validation of Chromatography Data Systems: Ensuring Data Integrity, Meeting Business and Regulatory Requirements Second Edition ed. (Royal Society of Chemistry, Cambridge, United Kingdom, 2017).
21.
B. Boehm, Some Information processing Implications of Air Force Missions: 1970–1980 (RAND Corporation, Santa Monica, California, 1970).
22.
S. Schmitt, personal communication, 2021.
23.
M. Frewster and D. Graham, Automated Software Testing - Effective Use of Test Execution Tools (Addison Wesley, Harlow, 1999).
24.
R.D.McDowall, Spectroscopy 36(4), 14–17 (2021).

